thermodynamics (from the Greek θέρμη therme, meaning "heat" and δύναμις, dynamis,energy conversion between heat and mechanical work, and subsequently the macroscopic variables such as temperature, volume and pressure. meaning force) is the study of

 Table of the thermodynamicists representative of the original eight founding schools of thermodynamics. The schools having the most-lasting effect in founding the modern versions of thermodynamics, being the Berlin school, particularly as established in Rudolf Clausius’s 1865 textbook The Mechanical Theory of Heat, which is the prototype of all modern textbooks on thermodynamics, the Vienna school, with the statistical mechanics of Ludwig Boltzmann, and the Gibbsian school at Yale University, American engineer Willard Gibbs' 1876 On the Equilibrium of Heterogeneous Substances launching chemical thermodynamics.
Table of the thermodynamicists representative of the original eight founding schools of thermodynamics. The schools having the most-lasting effect in founding the modern versions of thermodynamics, being the Berlin school, particularly as established in Rudolf Clausius’s 1865 textbook The Mechanical Theory of Heat, which is the prototype of all modern textbooks on thermodynamics, the Vienna school, with the statistical mechanics of Ludwig Boltzmann, and the Gibbsian school at Yale University, American engineer Willard Gibbs' 1876 On the Equilibrium of Heterogeneous Substances launching chemical thermodynamics.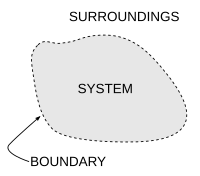
A diagram of a generic thermodynamic system
Historically, thermodynamics developed out of a need to increase the efficiency of early steam engines, particularly through the work of French physicist Nicolas Léonard Sadi Carnot (1824) who believed that engine efficiency was the key that could help France win the Napoleonic Wars. The first to give a concise definition of the subject was Scottish physicist William Thomson who in 1854 stated that:
Thermo-dynamics is the subject of the relation of heat to forces acting between contiguous parts of bodies, and the relation of heat to electrical agency.
Two fields of thermodynamics emerged in the following decades. Statistical thermodynamics, or statistical mechanics, (1860) concerned itself with statistical predictions of the collective motion of particles from their microscopic behavior, while chemical thermodynamics (1873) studies the nature of the role of entropy in the process of chemical reaction
Introduction
The starting point for most thermodynamic considerations are the laws of thermodynamics, which postulate that energy can be exchanged between physical systems as heat or work. They also postulate the existence of a quantity named entropy, which can be defined for any isolated system that is in thermodynamic equilibrium.
In thermodynamics, interactions between large ensembles of objects are studied and categorized. Central to this are the concepts of system and surroundings. A system is composed of particles, whose average motions define its properties, which in turn are related to one another through equations of state. Properties can be combined to express internal energy and thermodynamic potentials, which are useful for determining conditions for equilibrium and spontaneous processes.
With these tools, thermodynamics can be used to describe how systems respond to changes in their environment. This can be applied to a wide variety of topics in science and engineering, such as engines, phase transitions, chemical reactions, transport phenomena, and even black holes. The results of thermodynamics are essential for other fields of physics and for chemistry, chemical engineering, aerospace engineering, mechanical engineering, cell biology, biomedical engineering, materials science, and economics, to name a few.
The present article is focused mainly on classical thermodynamics, which is concerned with systems in thermodynamic equilibrium. It is wise to distinguish classical thermodynamics from non-equilibrium thermodynamics, which is concerned with systems that are not in thermodynamic equilibrium.
History
The history of thermodynamics as a scientific discipline generally begins with Otto von Guericke who, in 1650, built and designed the world's first vacuum pump and demonstrated a vacuum using his Magdeburg hemispheres. Guericke was driven to make a vacuum in order to disprove Aristotle's long-held supposition that 'nature abhors a vacuum'. Shortly after Guericke, the English physicist and chemist Robert Boyle had learned of Guericke's designs and, in 1656, in coordination with English scientist Robert Hooke, built an air pump.Using this pump, Boyle and Hooke noticed a correlation between pressure, temperature, and volume. In time, Boyle's Law was formulated, which states that pressure and volume are inversely proportional. Then, in 1679, based on these concepts, an associate of Boyle's named Denis Papin built a bone digester, which was a closed vessel with a tightly fitting lid that confined steam until a high pressure was generated.
Later designs implemented a steam release valve that kept the machine from exploding. By watching the valve rhythmically move up and down, Papin conceived of the idea of a piston and a cylinder engine. He did not, however, follow through with his design. Nevertheless, in 1697, based on Papin's designs, engineer Thomas Savery built the first engine, followed by Thomas Newcomen in 1712. Although these early engines were crude and inefficient, they attracted the attention of the leading scientists of the time. Their work led 127 years later to Sadi Carnot, the "father of thermodynamics", who, in 1824, published Reflections on the Motive Power of Fire, a discourse on heat, power, and engine efficiency. The paper outlined the basic energetic relations between the Carnot engine, the Carnot cycle, and motive power. It marked the start of thermodynamics as a modern science.
The term thermodynamics was coined by James Joule in 1849 to designate the science of relations between heat and power. By 1858, "thermo-dynamics", as a functional term, was used in William Thomson's paper An Account of Carnot's Theory of the Motive Power of Heat. The first thermodynamic textbook was written in 1859 by William Rankine, originally trained as a physicist and a civil and mechanical engineering professor at the University of Glasgow. The first and second laws of thermodynamics emerged simultaneously in the 1850s, primarily out of the works of William Rankine, Rudolf Clausius, and William Thomson (Lord Kelvin).
The foundations of statistical thermodynamics were set out by physicists such as James Clerk Maxwell, Ludwig Boltzmann, Max Planck, Rudolf Clausius and J. Willard Gibbs.
During the years 1873-76 the American mathematical physicist Josiah Willard Gibbs published a series of three papers, the most famous being On the Equilibrium of Heterogeneous Substances, in which he showed how thermodynamic processes could be graphically analyzed, by studying the energy, entropy, volume, temperature and pressure of the thermodynamic system in such a manner, one can determine if a process would occur spontaneously. During the early 20th century, chemists such as Gilbert N. Lewis, Merle Randall, and E. A. Guggenheim began to apply the mathematical methods of Gibbs to the analysis of chemical processes.
Interpretations
Thermodynamics has developed into several related branches of science, each with a different focus.
Classical thermodynamics
Classical thermodynamics is concerned with macroscopic thermodynamic states and properties of large, near-equilibrium systems. It is used to model exchanges of energy, work and heat based on the laws of thermodynamics. The term "classical" reflects the fact that it represents the level of knowledge in the early 1800s. An atomic interpretation of these principles was provided later by the development of statistical mechanics. Nonetheless, classical thermodynamics is still a practical and widely-used science.
Statistical mechanics
Statistical mechanics (or statistical thermodynamics) emerged only with the development of atomic and molecular theories in the late 1800s and early 1900s, giving thermodynamics a molecular interpretation. This field relates the microscopic properties of individual atoms and molecules to the macroscopic or bulk properties of materials that can be observed in everyday life, thereby explaining thermodynamics as a natural result of statistics and mechanics (classical and quantum) at the microscopic level. This statistical approach is in contrast to classical thermodynamics, which is a more phenomenological approach.
Chemical thermodynamics
Chemical thermodynamics is the study of the interrelation of energy with chemical reactions or with a physical change of state within the confines of the laws of thermodynamics.
Treatment of equilibrium
Equilibrium thermodynamics is the systematic study of transformations of matter and energy in systems as they approach equilibrium. The word equilibrium implies a state of balance. In an equilibrium state there are no unbalanced potentials, or driving forces, within the system. A central aim in equilibrium thermodynamics is: given a system in a well-defined initial state, subject to accurately specified constraints, to calculate what the state of the system will be once it has reached equilibrium.
Non-equilibrium thermodynamics is a branch of thermodynamics that deals with systems that are not in thermodynamic equilibrium. Most systems found in nature are not in thermodynamic equilibrium because they are not in stationary states, and are continuously and discontinuously subject to flux of matter and energy to and from other systems. The thermodynamic study of non-equilibrium systems requires more general concepts than are dealt with by equilibrium thermodynamics. Many natural systems still today remain beyond the scope of currently known macroscopic thermodynamic methods.
Laws of thermodynamics
Thermodynamics defines four laws which do not depend on the details of the systems under study or how they interact. Hence these laws are generally valid, can be applied to systems about which one knows nothing other than the balance of energy and matter transfer. Examples of such systems include Einstein's prediction of spontaneous emission, and ongoing research into the thermodynamics of black holes.
These four laws are:
- Zeroth law of thermodynamics: If two systems are in thermal equilibrium with a third, they are also in thermal equilibrium with each other.
Systems are said to be in equilibrium if the small, random exchanges (due to Brownian motion, for example) between them do not lead to a net change in the total energy summed over all systems. This law is tacitly assumed in every measurement of temperature. Thus, if we want to know if two bodies are at the same temperature, it is not necessary to bring them into contact and to watch whether their observable properties change with time.
This law was considered so obvious it was added as a virtual afterthought, hence the designation Zeroth, rather than Fourth. In short, if the temperature of material A is equal to the temperature of material B, and B is equal to the temperature of material C, then A and C must also be equal. This implies that thermal equilibrium is an equivalence relation on the set of thermodynamic systems.
- First law of thermodynamics: The internal energy of an isolated system is constant.
The first law of thermodynamics, an expression of the principle of conservation of energy, states that energy can be transformed (changed from one form to another), but cannot be created or destroyed.
It is usually formulated by saying that the change in the internal energy of a closed thermodynamic system is equal to the amount of heat supplied to the system, minus the amount of work done by the system on its surroundings. Work and heat are due to processes which add or subtract energy, while internal energy is a particular form of energy associated with the system. Internal energy is a property of the system whereas work done and heat supplied are not. A significant result of this distinction is that a given internal energy change can be achieved by many combinations of heat and work.
- Second law of thermodynamics: Heat cannot spontaneously flow from a colder location to a hotter location.
The second law of thermodynamics is an expression of the universal principle of decay observable in nature. The second law is an observation of the fact that over time, differences in temperature, pressure, and chemical potential tend to even out in a physical system that is isolated from the outside world. Entropy is a measure of how much this evening-out process has progressed. The entropy of an isolated system which is not in equilibrium will tend to increase over time, approaching a maximum value at equilibrium.
In classical thermodynamics, the second law is a basic postulate applicable to any system involving heat energy transfer; in statistical thermodynamics, the second law is a consequence of the assumed randomness of molecular chaos. There are many versions of the second law, but they all have the same effect, which is to explain the phenomenon of irreversibility in nature.
- Third law of thermodynamics: As a system approaches absolute zero, all processes cease and the entropy of the system approaches a minimum value.
The third law of thermodynamics is a statistical law of nature regarding entropy and the impossibility of reaching absolute zero of temperature. This law provides an absolute reference point for the determination of entropy. The entropy determined relative to this point is the absolute entropy. Alternate definitions are, "the entropy of all systems and of all states of a system is smallest at absolute zero," or equivalently "it is impossible to reach the absolute zero of temperature by any finite number of processes".
Absolute zero, at which all activity would stop if it were possible to happen, is −273.15 °C (degrees Celsius), or −459.67 °F (degrees Fahrenheit) or 0 K (kelvin).
System models
An important concept in thermodynamics is the thermodynamic system, a precisely defined region of the universe under study. Everything in the universe except the system is known as the surroundings. A system is separated from the remainder of the universe by a boundary which may be notional or not, but which by convention delimits a finite volume. Exchanges of work, heat, or matter between the system and the surroundings take place across this boundary.
In practice, the boundary is simply an imaginary dotted line drawn around a volume when there is going to be a change in the internal energy of that volume. Anything that passes across the boundary that effects a change in the internal energy needs to be accounted for in the energy balance equation. The volume can be the region surrounding a single atom resonating energy, such as Max Planck defined in 1900; it can be a body of steam or air in a steam engine, such as Sadi Carnot defined in 1824; it can be the body of a tropical cyclone, such as Kerry Emanuel theorized in 1986 in the field of atmospheric thermodynamics; it could also be just one nuclidequarks) as hypothesized in quantum thermodynamics. (i.e. a system of
Boundaries are of four types: fixed, moveable, real, and imaginary. For example, in an engine, a fixed boundary means the piston is locked at its position; as such, a constant volume process occurs. In that same engine, a moveable boundary allows the piston to move in and out. For closed systems, boundaries are real while for open system boundaries are often imaginary.
Generally, thermodynamics distinguishes five classes of systems, defined in terms of what is allowed to cross their boundaries:
| Type of system | Allows Matter | Allows Work | Allows Heat |
|---|---|---|---|
| Open | |||
| Closed | |||
| Isolated |
As time passes in an isolated system, internal differences in the system tend to even out and pressures and temperatures tend to equalize, as do density differences. A system in which all equalizing processes have gone to completion is considered to be in a state of thermodynamic equilibrium.
In thermodynamic equilibrium, a system's properties are, by definition, unchanging in time. Systems in equilibrium are much simpler and easier to understand than systems which are not in equilibrium. Often, when analysing a thermodynamic process, it can be assumed that each intermediate state in the process is at equilibrium. This will also considerably simplify the situation. Thermodynamic processes which develop so slowly as to allow each intermediate step to be an equilibrium state are said to be reversible processes.
States and processes
When a system is at equilibrium under a given set of conditions, it is said to be in a definite thermodynamic state. The state of the system can be described by a number of intensive variables and extensive variables. The properties of the system can be described by an equation of state which specifies the relationship between these variables. State may be thought of as the instantaneous quantitative description of a system with a set number of variables held constant.
A thermodynamic process may be defined as the energetic evolution of a thermodynamic system proceeding from an initial state to a final state. Typically, each thermodynamic process is distinguished from other processes in energetic character according to what parameters, such as temperature, pressure, or volume, etc., are held fixed. Furthermore, it is useful to group these processes into pairs, in which each variable held constant is one member of a conjugate pair.
Several common thermodynamic processes are:
- Isobaric process: occurs at constant pressure
- Isochoric process: occurs at constant volume (also called isometric/isovolumetric)
- Isothermal process: occurs at a constant temperature
- Adiabatic process: occurs without loss or gain of energy by heat
- Isentropic process: a reversible adiabatic process, occurs at a constant entropy
- Isenthalpic process: occurs at a constant enthalpy
- Steady state process: occurs without a change in the internal energy
Instrumentation
There are two types of thermodynamic instruments, the meter and the reservoir. A thermodynamic meter is any device which measures any parameter of a thermodynamic system. In some cases, the thermodynamic parameter is actually defined in terms of an idealized measuring instrument. For example, the zeroth law states that if two bodies are in thermal equilibrium with a third body, they are also in thermal equilibrium with each other. This principle, as noted by James Maxwell in 1872, asserts that it is possible to measure temperature. An idealized thermometer is a sample of an ideal gas at constant pressure. From the ideal gas law pV=nRT, the volume of such a sample can be used as an indicator of temperature; in this manner it defines temperature. Although pressure is defined mechanically, a pressure-measuring device, called a barometer may also be constructed from a sample of an ideal gas held at a constant temperature. A calorimeter is a device which is used to measure and define the internal energy of a system.
A thermodynamic reservoir is a system which is so large that it does not appreciably alter its state parameters when brought into contact with the test system. It is used to impose a particular value of a state parameter upon the system. For example, a pressure reservoir is a system at a particular pressure, which imposes that pressure upon any test system that it is mechanically connected to. The Earth's atmosphere is often used as a pressure reservoir.
Conjugate variables
The central concept of thermodynamics is that of energy, the ability to do work. By the First Law, the total energy of a system and its surroundings is conserved. Energy may be transferred into a system by heating, compression, or addition of matter, and extracted from a system by cooling, expansion, or extraction of matter. In mechanics, for example, energy transfer equals the product of the force applied to a body and the resulting displacement.
Conjugate variables are pairs of thermodynamic concepts, with the first being akin to a "force" applied to some thermodynamic system, the second being akin to the resulting "displacement," and the product of the two equalling the amount of energy transferred. The common conjugate variables are:
- Pressure-volume (the mechanical parameters);
- Temperature-entropy (thermal parameters);
- Chemical potential-particle number (material parameters).
Potentials
Thermodynamic potentials are different quantitative measures of the stored energy in a system. Potentials are used to measure energy changes in systems as they evolve from an initial state to a final state. The potential used depends on the constraints of the system, such as constant temperature or pressure. For example, the Helmholtz and Gibbs energies are the energies available in a system to do useful work when the temperature and volume or the pressure and temperature are fixed, respectively.
The five most well known potentials are:
| Name | Symbol | Formula | Natural variables |
|---|---|---|---|
| Internal energy | U | TS − pV + 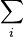 μiNi μiNi | S,V,{Ni} |
| Helmholtz free energy | F, A | U − TS | T,V,{Ni} |
| Enthalpy | H | U + pV | S,p,{Ni} |
| Gibbs free energy | G | U + pV − TS | T,p,{Ni} |
| Landau Potential (Grand potential) | Ω, ΦG | U − TS − μiNi | T,V,{μi} |
where T is the temperature, S the entropy, p the pressure, V the volume, μ the chemical potential, N the number of particles in the system, and i is the count of particles types in the system.
Thermodynamic potentials can be derived from the energy balance equation applied to a thermodynamic system. Other thermodynamic potentials can also be obtained through Legendre transformation.







{kind=link}